---
title: "Differentiation Progression Analysis"
subtitle: >
Quantifying differentiation timing differences
from bulk RNA-seq time course data
format:
html:
code-fold: true
code-tools: true
toc: true
toc-depth: 3
fig-width: 8
fig-height: 6
fig-dpi: 150
execute:
warning: false
message: false
---
## Overview
PCA-based method for comparing differentiation
progression between conditions in bulk RNA-seq time
course data.
**Dataset.** Data from
[Martinez et al. (2024)](https://doi.org/10.3389/fncel.2024.1341141):
D21 (euploid control) and T21 (trisomy 21)
iPSC-derived neural progenitors at Days 6, 10,
and 17 (18 samples, 3 replicates per condition).
## Workflow
{width="55%"
fig-align="center"}
## Mathematical Framework
### Maturation Score
Each sample is projected onto $\vec{v}$ and
normalized so the early centroid maps to 0 and the
late centroid maps to 1:
$$
s_i
= \frac{
\left(\mathbf{z}_i
- \bar{\mathbf{z}}_{t_{\textrm{early}}}\right)
\cdot \vec{v}
}{
\lVert\vec{v}\rVert^2
}
$$
---
## Analysis
### Setup
```{r}
#| label: setup
library(DESeq2)
library(ggplot2)
library(dplyr)
library(tidyr)
library(tibble)
library(plotly)
library(ComplexHeatmap)
library(circlize)
library(viridis)
meta <- readRDS("dat/metadata/WC24_metadata_clean.rds")
raw_counts <- readRDS(
"dat/counts/raw/WC24_filt_raw_counts.rds"
)
vst_counts <- readRDS(
"dat/counts/vst/WC24_vst_counts.rds"
)
meta$timepoint <- factor(
meta$timepoint, levels = c(6, 10, 17)
)
theme_pub <- function(base_size = 10) {
theme_classic(base_size = base_size) +
theme(
text = element_text(color = "black"),
axis.text = element_text(
size = base_size - 2, color = "black"
),
axis.title = element_text(
size = base_size - 1, color = "black"
),
axis.line = element_line(
color = "black", linewidth = 0.3
),
axis.ticks = element_line(
linewidth = 0.3, color = "black"
),
panel.grid.major = element_line(
color = "#D9D9D9", linewidth = 0.22
),
panel.grid.minor = element_blank(),
legend.title = element_blank(),
legend.text = element_text(
size = base_size - 2, color = "black"
),
legend.key.size = unit(0.35, "cm"),
strip.text = element_text(
size = base_size - 1, face = "bold",
color = "black"
),
strip.background = element_blank()
)
}
col_d21 <- "#1F78B4"
col_t21 <- "#E31A1C"
```
### Feature Selection: Likelihood Ratio Test
```{r}
#| label: lrt
#| cache: true
ctrl_meta <- meta[meta$genotype == "D21", ]
ctrl_counts <- raw_counts[, ctrl_meta$sample_id]
dds <- DESeqDataSetFromMatrix(
countData = ctrl_counts,
colData = ctrl_meta,
design = ~ timepoint
)
dds <- DESeq(dds, test = "LRT", reduced = ~ 1)
res <- results(dds)
res <- res[order(res$padj), ]
sig_genes <- rownames(res)[
!is.na(res$padj) & res$padj < 0.05
]
n_top <- min(1000, length(sig_genes))
top_genes <- head(rownames(res), n_top)
cat(
"Significant genes (padj < 0.05):",
length(sig_genes), "\n",
"Genes used for PCA:", n_top
)
```
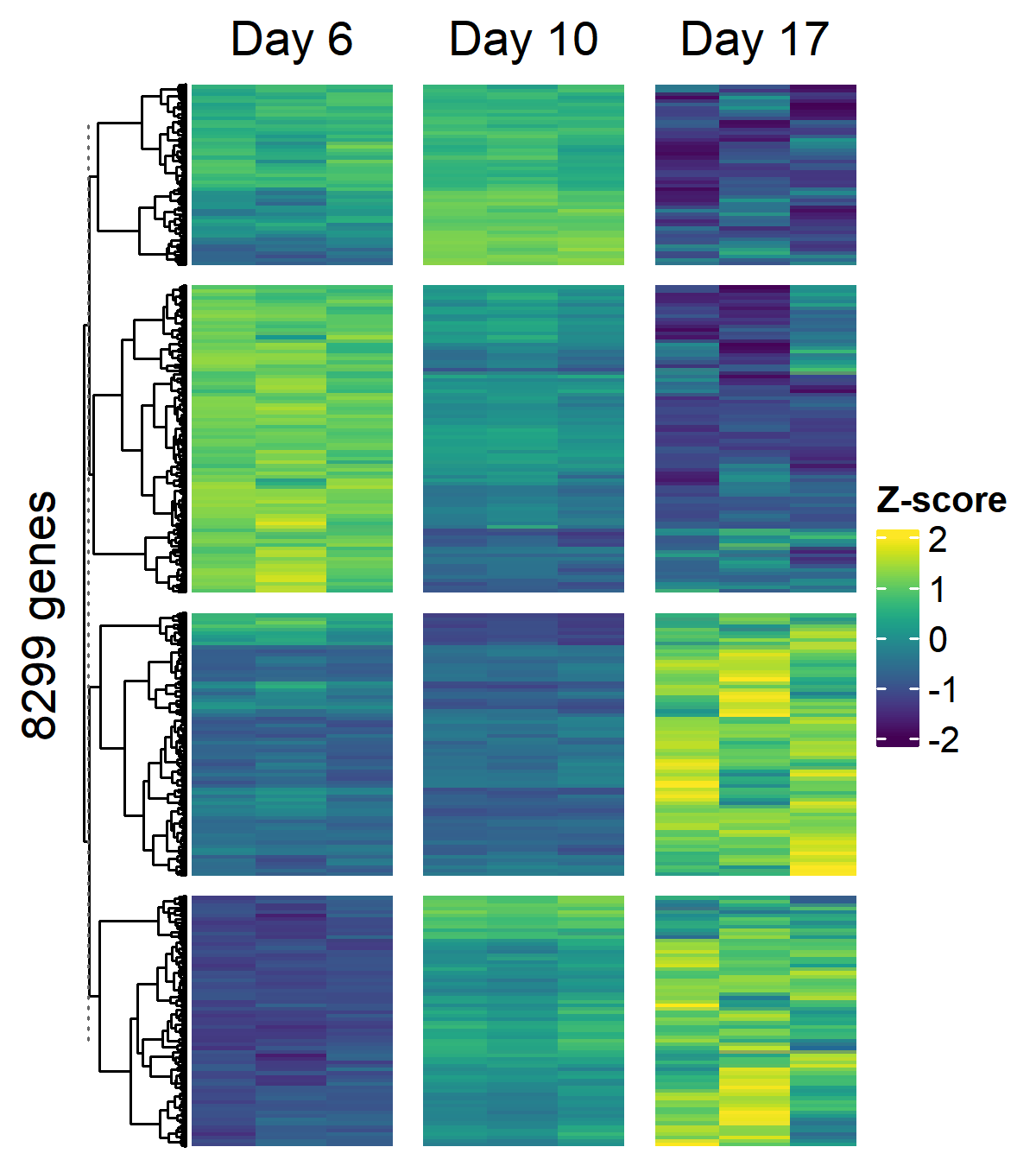
### Temporally Variable Gene Heatmap
```{r}
#| label: fig-heatmap
#| fig-cap: >
#| Z-scored expression of temporally variable genes
#| across D21 control timepoints (k=4 row clusters,
#| viridis palette, winsorized at +/- 2)
#| fig-height: 4.5
#| fig-width: 4
hm_mat <- as.matrix(
vst_counts[sig_genes, ctrl_meta$sample_id]
)
hm_z <- t(scale(t(hm_mat)))
hm_z[hm_z > 2] <- 2
hm_z[hm_z < -2] <- -2
col_fun <- colorRamp2(
seq(-2, 2, length.out = 100),
viridis(100)
)
ht <- Heatmap(
hm_z,
name = "Z-score",
col = col_fun,
cluster_columns = FALSE,
cluster_rows = TRUE,
clustering_method_rows = "ward.D2",
row_km = 4,
row_gap = unit(2, "mm"),
show_row_names = FALSE,
show_column_names = FALSE,
column_split = factor(
ctrl_meta[colnames(hm_z), "timepoint"],
levels = c(6, 10, 17)
),
column_title = c("Day 6", "Day 10", "Day 17"),
column_gap = unit(3, "mm"),
row_title = paste0(
length(sig_genes), " genes"
),
border = FALSE,
use_raster = TRUE
)
draw(ht)
```
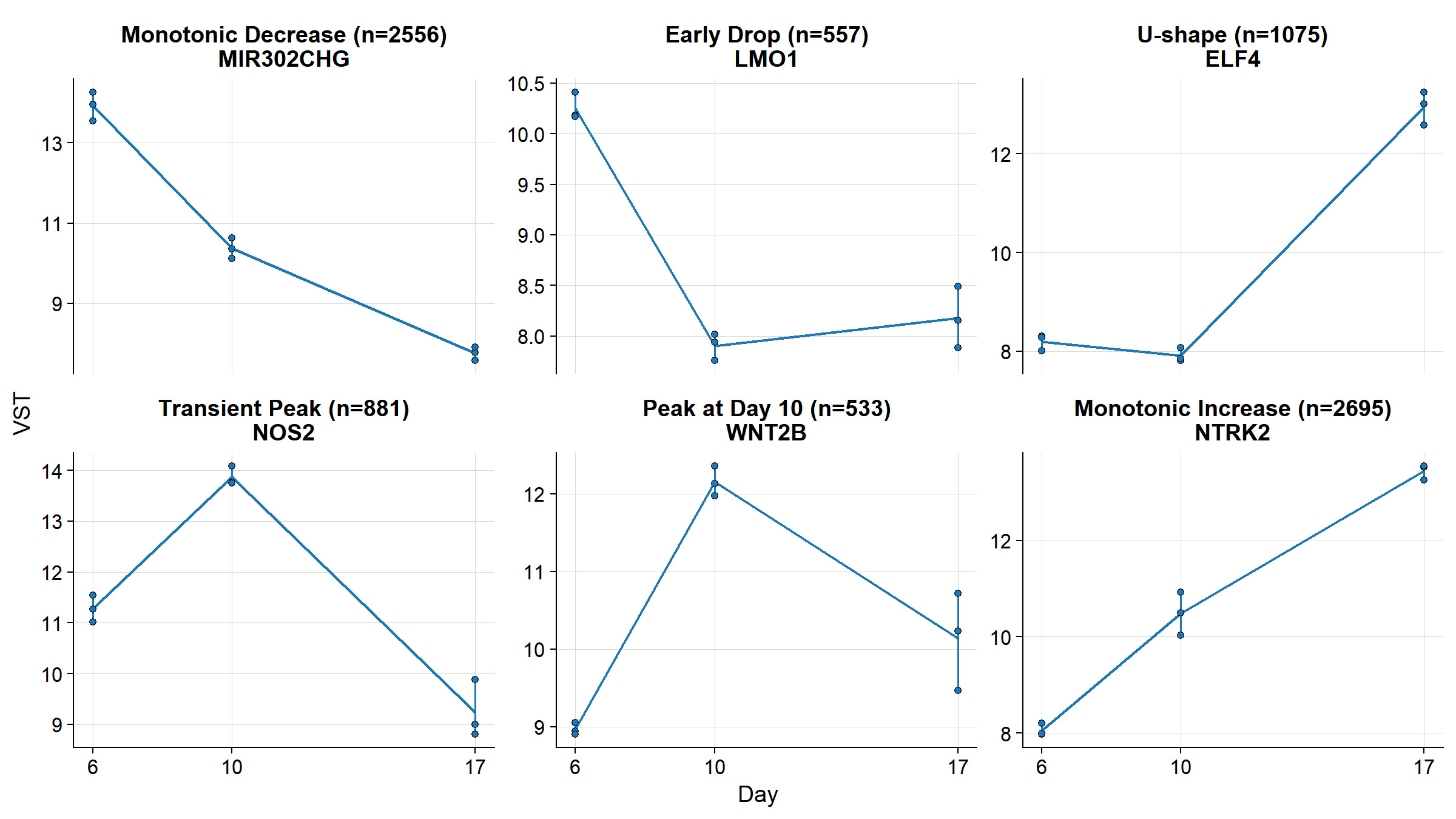
### Temporal Expression Patterns
Six temporal profiles identified among
LRT-significant genes.
```{r}
#| label: fig-patterns
#| fig-cap: >
#| Six temporal expression patterns identified
#| by the LRT. Each panel shows the gene with the
#| largest dynamic range for that pattern class.
#| Points = replicates, line = mean, bars = range.
#| fig-height: 4.5
#| fig-width: 8
ctrl_vst <- vst_counts[sig_genes, ctrl_meta$sample_id]
tp_num <- as.numeric(
as.character(ctrl_meta$timepoint)
)
mean_d6 <- rowMeans(ctrl_vst[, tp_num == 6])
mean_d10 <- rowMeans(ctrl_vst[, tp_num == 10])
mean_d17 <- rowMeans(ctrl_vst[, tp_num == 17])
# All 6 possible 3-timepoint orderings
patterns <- list(
"Monotonic Decrease" = names(which(
mean_d6 > mean_d10 & mean_d10 > mean_d17
)),
"Early Drop" = names(which(
mean_d6 > mean_d17 & mean_d17 > mean_d10
)),
"U-shape" = names(which(
mean_d17 > mean_d6 & mean_d6 > mean_d10
)),
"Transient Peak" = names(which(
mean_d10 > mean_d6 & mean_d6 > mean_d17
)),
"Peak at Day 10" = names(which(
mean_d10 > mean_d17 & mean_d17 > mean_d6
)),
"Monotonic Increase" = names(which(
mean_d17 > mean_d10 & mean_d10 > mean_d6
))
)
dyn_range <- apply(
cbind(mean_d6, mean_d10, mean_d17), 1,
function(x) max(x) - min(x)
)
# Pick top gene per pattern
example_genes <- vapply(
names(patterns), function(nm) {
genes <- patterns[[nm]]
if (length(genes) == 0) return(NA_character_)
genes[which.max(dyn_range[genes])]
}, character(1)
)
# Count genes per pattern
pattern_counts <- vapply(
patterns, length, integer(1)
)
# Keep only patterns with genes
keep <- !is.na(example_genes)
example_genes <- example_genes[keep]
pattern_names <- names(example_genes)
# Build plot data
plot_df <- do.call(rbind, lapply(
seq_along(example_genes), function(i) {
g <- example_genes[i]
nm <- pattern_names[i]
data.frame(
gene = g,
pattern = nm,
n_genes = pattern_counts[nm],
timepoint = tp_num,
expr = as.numeric(ctrl_vst[g, ]),
sample = ctrl_meta$sample_id
)
}
))
plot_df$label <- paste0(
plot_df$pattern,
" (n=", plot_df$n_genes, ")",
"\n", plot_df$gene
)
plot_df$label <- factor(
plot_df$label, levels = unique(plot_df$label)
)
plot_means <- plot_df %>%
group_by(label, timepoint) %>%
summarize(
m = mean(expr),
ymin = min(expr),
ymax = max(expr),
.groups = "drop"
)
ggplot() +
geom_line(
data = plot_means,
aes(x = timepoint, y = m),
color = col_d21, linewidth = 0.55
) +
geom_linerange(
data = plot_means,
aes(x = timepoint, ymin = ymin,
ymax = ymax),
linewidth = 0.35, color = col_d21
) +
geom_point(
data = plot_df,
aes(x = timepoint, y = expr),
shape = 21, size = 1.2,
stroke = 0.25, color = "black",
fill = col_d21, alpha = 1
) +
facet_wrap(~ label, nrow = 2,
scales = "free_y") +
scale_x_continuous(
breaks = c(6, 10, 17),
labels = c("6", "10", "17")
) +
labs(x = "Day", y = "VST") +
theme_pub(base_size = 10)
```
### D21 Control PC Space
```{r}
#| label: pca-compute
pca_input <- t(as.matrix(
vst_counts[top_genes, ctrl_meta$sample_id]
))
pca_res <- prcomp(pca_input, scale. = TRUE,
center = TRUE)
var_pct <- round(
summary(pca_res)$importance[2, 1:3] * 100, 1
)
all_input <- t(as.matrix(
vst_counts[top_genes, meta$sample_id]
))
all_proj <- predict(pca_res, all_input)
pca_df <- data.frame(
PC1 = all_proj[, 1],
PC2 = all_proj[, 2],
PC3 = all_proj[, 3],
sample = meta$sample_id,
genotype = meta$genotype,
timepoint = factor(
paste0("Day ", meta$timepoint),
levels = c("Day 6", "Day 10", "Day 17")
)
)
tp_num_all <- as.numeric(
as.character(meta$timepoint)
)
ctrl_d6 <- meta$genotype == "D21" & tp_num_all == 6
ctrl_d10 <- meta$genotype == "D21" & tp_num_all == 10
ctrl_d17 <- meta$genotype == "D21" & tp_num_all == 17
centroid_d6 <- colMeans(all_proj[ctrl_d6, 1:3])
centroid_d10 <- colMeans(all_proj[ctrl_d10, 1:3])
centroid_d17 <- colMeans(all_proj[ctrl_d17, 1:3])
tp_colors <- c(
"Day 6" = "#2563EB",
"Day 10" = "#059669",
"Day 17" = "#DC2626"
)
geno_symbols <- c(
"D21" = "circle", "T21" = "diamond"
)
scene_axes <- list(
xaxis = list(title = paste0(
"PC1 (", var_pct[1], "%)"
)),
yaxis = list(title = paste0(
"PC2 (", var_pct[2], "%)"
)),
zaxis = list(title = paste0(
"PC3 (", var_pct[3], "%)"
))
)
# Curved arc through the 3 centroids
t_param <- c(0, 0.5, 1)
t_interp <- seq(0, 1, length.out = 40)
arc_x <- spline(
t_param,
c(centroid_d6[1], centroid_d10[1],
centroid_d17[1]),
xout = t_interp
)$y
arc_y <- spline(
t_param,
c(centroid_d6[2], centroid_d10[2],
centroid_d17[2]),
xout = t_interp
)$y
arc_z <- spline(
t_param,
c(centroid_d6[3], centroid_d10[3],
centroid_d17[3]),
xout = t_interp
)$y
arc_dir <- c(
arc_x[40] - arc_x[39],
arc_y[40] - arc_y[39],
arc_z[40] - arc_z[39]
)
```
```{r}
#| label: fig-d21-pca
#| fig-cap: >
#| D21 control samples in PC space (top 1000 LRT
#| genes). Curved line traces the differentiation
#| arc through timepoint centroids.
ctrl_pca <- pca_df[pca_df$genotype == "D21", ]
plot_ly() %>%
add_trace(
data = ctrl_pca,
x = ~PC1, y = ~PC2, z = ~PC3,
color = ~timepoint,
colors = tp_colors,
type = "scatter3d", mode = "markers",
marker = list(size = 4, opacity = 1),
text = ~paste(sample, timepoint),
hoverinfo = "text"
) %>%
add_trace(
x = arc_x, y = arc_y, z = arc_z,
type = "scatter3d", mode = "lines",
line = list(
width = 5, color = "#000", dash = "solid"
),
name = "Differentiation Arc",
showlegend = TRUE
) %>%
add_trace(
type = "cone",
x = arc_x[40], y = arc_y[40], z = arc_z[40],
u = arc_dir[1], v = arc_dir[2],
w = arc_dir[3],
sizemode = "absolute", sizeref = 2,
anchor = "tail", showscale = FALSE,
colorscale = list(c(0, "#000"), c(1, "#000")),
name = "", showlegend = FALSE
) %>%
# Labels at centroids
add_trace(
x = centroid_d6[1], y = centroid_d6[2],
z = centroid_d6[3],
type = "scatter3d", mode = "text",
text = "Start",
textfont = list(
size = 12, color = "#000", family = "Arial"
),
textposition = "top center",
showlegend = FALSE, name = ""
) %>%
add_trace(
x = centroid_d10[1], y = centroid_d10[2],
z = centroid_d10[3],
type = "scatter3d", mode = "text",
text = "Middle",
textfont = list(
size = 12, color = "#000", family = "Arial"
),
textposition = "top center",
showlegend = FALSE, name = ""
) %>%
add_trace(
x = centroid_d17[1], y = centroid_d17[2],
z = centroid_d17[3],
type = "scatter3d", mode = "text",
text = "End",
textfont = list(
size = 12, color = "#000", family = "Arial"
),
textposition = "top center",
showlegend = FALSE, name = ""
) %>%
layout(scene = scene_axes)
```
### Choosing the Reference Vector
Three possible centroid-to-centroid vectors, each
capturing a different segment of the trajectory.
```{r}
#| label: fig-vector-options
#| fig-cap: >
#| Three candidate reference vectors. The full
#| trajectory (Day 6 to Day 17, green solid) is
#| selected because it spans the complete
#| differentiation arc.
vec_early <- centroid_d10 - centroid_d6
vec_late <- centroid_d17 - centroid_d10
vec_full <- centroid_d17 - centroid_d6
mid_early <- (centroid_d6 + centroid_d10) / 2
mid_late <- (centroid_d10 + centroid_d17) / 2
mid_full <- (centroid_d6 + centroid_d17) / 2
plot_ly() %>%
add_trace(
data = ctrl_pca,
x = ~PC1, y = ~PC2, z = ~PC3,
color = ~timepoint,
colors = tp_colors,
type = "scatter3d", mode = "markers",
marker = list(size = 4, opacity = 1),
text = ~paste(sample, timepoint),
hoverinfo = "text"
) %>%
# D6 -> D10
add_trace(
x = c(centroid_d6[1], centroid_d10[1]),
y = c(centroid_d6[2], centroid_d10[2]),
z = c(centroid_d6[3], centroid_d10[3]),
type = "scatter3d", mode = "lines",
line = list(width = 5, color = "#6366F1"),
name = "D6 -> D10"
) %>%
add_trace(
type = "cone",
x = centroid_d10[1], y = centroid_d10[2],
z = centroid_d10[3],
u = vec_early[1], v = vec_early[2],
w = vec_early[3],
sizemode = "absolute", sizeref = 2,
anchor = "tail", showscale = FALSE,
colorscale = list(
c(0, "#6366F1"), c(1, "#6366F1")
),
showlegend = FALSE, name = ""
) %>%
add_trace(
x = mid_early[1], y = mid_early[2],
z = mid_early[3],
type = "scatter3d", mode = "text",
text = "D6->D10",
textfont = list(size = 10, color = "#6366F1"),
showlegend = FALSE, name = ""
) %>%
# D10 -> D17
add_trace(
x = c(centroid_d10[1], centroid_d17[1]),
y = c(centroid_d10[2], centroid_d17[2]),
z = c(centroid_d10[3], centroid_d17[3]),
type = "scatter3d", mode = "lines",
line = list(width = 5, color = "#F59E0B"),
name = "D10 -> D17"
) %>%
add_trace(
type = "cone",
x = centroid_d17[1], y = centroid_d17[2],
z = centroid_d17[3],
u = vec_late[1], v = vec_late[2],
w = vec_late[3],
sizemode = "absolute", sizeref = 2,
anchor = "tail", showscale = FALSE,
colorscale = list(
c(0, "#F59E0B"), c(1, "#F59E0B")
),
showlegend = FALSE, name = ""
) %>%
add_trace(
x = mid_late[1], y = mid_late[2],
z = mid_late[3],
type = "scatter3d", mode = "text",
text = "D10->D17",
textfont = list(size = 10, color = "#F59E0B"),
showlegend = FALSE, name = ""
) %>%
# D6 -> D17
add_trace(
x = c(centroid_d6[1], centroid_d17[1]),
y = c(centroid_d6[2], centroid_d17[2]),
z = c(centroid_d6[3], centroid_d17[3]),
type = "scatter3d", mode = "lines",
line = list(width = 7, color = "#059669"),
name = "D6 -> D17"
) %>%
add_trace(
type = "cone",
x = centroid_d17[1] + vec_late[1] * 0.01,
y = centroid_d17[2] + vec_late[2] * 0.01,
z = centroid_d17[3] + vec_late[3] * 0.01,
u = vec_full[1], v = vec_full[2],
w = vec_full[3],
sizemode = "absolute", sizeref = 2.5,
anchor = "tail", showscale = FALSE,
colorscale = list(
c(0, "#059669"), c(1, "#059669")
),
showlegend = FALSE, name = ""
) %>%
add_trace(
x = mid_full[1], y = mid_full[2],
z = mid_full[3],
type = "scatter3d", mode = "text",
text = "D6->D17",
textfont = list(size = 11, color = "#059669"),
showlegend = FALSE, name = ""
) %>%
layout(scene = scene_axes)
```
### All Samples in Reference PC Space
```{r}
#| label: fig-all-pca
#| fig-cap: >
#| All D21 and T21 samples projected into the
#| D21-defined PC space (color = timepoint,
#| shape = genotype)
plot_ly(
pca_df,
x = ~PC1, y = ~PC2, z = ~PC3,
color = ~timepoint,
symbol = ~genotype,
colors = tp_colors,
symbols = geno_symbols,
type = "scatter3d",
mode = "markers",
marker = list(size = 4, opacity = 1),
text = ~paste(sample, genotype, timepoint),
hoverinfo = "text"
) %>%
layout(scene = scene_axes)
```
### Reference Trajectory
```{r}
#| label: fig-ref-trajectory
#| fig-cap: >
#| Reference trajectory (green) from D21 Day 6
#| to Day 17 centroid with arrowhead.
#| Black diamonds = centroids.
ref_vec <- centroid_d17 - centroid_d6
plot_ly() %>%
add_trace(
data = pca_df,
x = ~PC1, y = ~PC2, z = ~PC3,
color = ~timepoint,
symbol = ~genotype,
colors = tp_colors,
symbols = geno_symbols,
type = "scatter3d", mode = "markers",
marker = list(size = 4, opacity = 1),
text = ~paste(sample, genotype, timepoint),
hoverinfo = "text"
) %>%
add_trace(
x = c(centroid_d6[1], centroid_d17[1]),
y = c(centroid_d6[2], centroid_d17[2]),
z = c(centroid_d6[3], centroid_d17[3]),
type = "scatter3d", mode = "lines",
line = list(width = 7, color = "#059669"),
name = "Reference Vector (D6->D17)",
showlegend = TRUE
) %>%
add_trace(
type = "cone",
x = centroid_d17[1], y = centroid_d17[2],
z = centroid_d17[3],
u = ref_vec[1] * 0.15,
v = ref_vec[2] * 0.15,
w = ref_vec[3] * 0.15,
sizemode = "absolute", sizeref = 3,
anchor = "tail", showscale = FALSE,
colorscale = list(
c(0, "#059669"), c(1, "#059669")
),
name = "", showlegend = FALSE
) %>%
# Cluster labels at centroids
add_trace(
x = centroid_d6[1], y = centroid_d6[2],
z = centroid_d6[3],
type = "scatter3d", mode = "text",
text = "Day 6",
textfont = list(
size = 11, color = tp_colors["Day 6"],
family = "Arial"
),
textposition = "top center",
showlegend = FALSE, name = ""
) %>%
add_trace(
x = centroid_d10[1], y = centroid_d10[2],
z = centroid_d10[3],
type = "scatter3d", mode = "text",
text = "Day 10",
textfont = list(
size = 11, color = tp_colors["Day 10"],
family = "Arial"
),
textposition = "top center",
showlegend = FALSE, name = ""
) %>%
add_trace(
x = centroid_d17[1], y = centroid_d17[2],
z = centroid_d17[3],
type = "scatter3d", mode = "text",
text = "Day 17",
textfont = list(
size = 11, color = tp_colors["Day 17"],
family = "Arial"
),
textposition = "top center",
showlegend = FALSE, name = ""
) %>%
layout(scene = scene_axes)
```
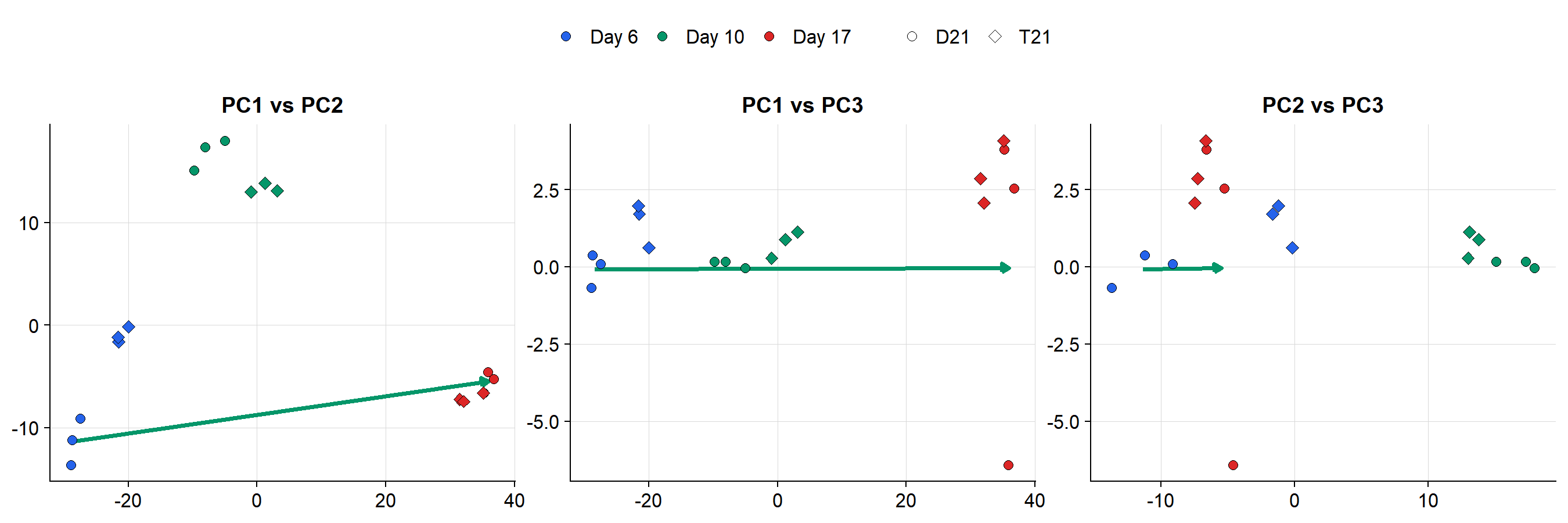
### 2D PC Projections with Reference Vector
```{r}
#| label: fig-pc-panels
#| fig-cap: >
#| Pairwise PC projections of all samples with
#| the D6-to-D17 reference vector (green arrow).
#| Shape = genotype, color = timepoint.
#| fig-height: 3
#| fig-width: 9
# Build segment data for the reference vector
vec_seg <- data.frame(
x = centroid_d6[1:3],
xend = centroid_d17[1:3],
panel = c("PC1 vs PC2", "PC1 vs PC3",
"PC2 vs PC3")
)
# Build panel data
panel_df <- rbind(
data.frame(
x = pca_df$PC1, y = pca_df$PC2,
panel = "PC1 vs PC2",
genotype = pca_df$genotype,
timepoint = pca_df$timepoint
),
data.frame(
x = pca_df$PC1, y = pca_df$PC3,
panel = "PC1 vs PC3",
genotype = pca_df$genotype,
timepoint = pca_df$timepoint
),
data.frame(
x = pca_df$PC2, y = pca_df$PC3,
panel = "PC2 vs PC3",
genotype = pca_df$genotype,
timepoint = pca_df$timepoint
)
)
panel_df$panel <- factor(
panel_df$panel,
levels = c("PC1 vs PC2", "PC1 vs PC3",
"PC2 vs PC3")
)
# Vector endpoints per panel
seg_df <- data.frame(
x = c(centroid_d6[1], centroid_d6[1],
centroid_d6[2]),
y = c(centroid_d6[2], centroid_d6[3],
centroid_d6[3]),
xend = c(centroid_d17[1], centroid_d17[1],
centroid_d17[2]),
yend = c(centroid_d17[2], centroid_d17[3],
centroid_d17[3]),
panel = factor(
c("PC1 vs PC2", "PC1 vs PC3",
"PC2 vs PC3"),
levels = c("PC1 vs PC2", "PC1 vs PC3",
"PC2 vs PC3")
)
)
# Axis labels per panel
ax_labels <- data.frame(
panel = factor(
c("PC1 vs PC2", "PC1 vs PC3",
"PC2 vs PC3"),
levels = c("PC1 vs PC2", "PC1 vs PC3",
"PC2 vs PC3")
),
xlab = c(
paste0("PC1 (", var_pct[1], "%)"),
paste0("PC1 (", var_pct[1], "%)"),
paste0("PC2 (", var_pct[2], "%)")
),
ylab = c(
paste0("PC2 (", var_pct[2], "%)"),
paste0("PC3 (", var_pct[3], "%)"),
paste0("PC3 (", var_pct[3], "%)")
)
)
ggplot(panel_df, aes(x = x, y = y)) +
geom_segment(
data = seg_df,
aes(x = x, y = y, xend = xend, yend = yend),
color = "#059669", linewidth = 0.8,
arrow = arrow(
length = unit(0.12, "cm"), type = "closed"
),
inherit.aes = FALSE
) +
geom_point(
aes(fill = timepoint, shape = genotype),
size = 1.8, stroke = 0.25, color = "black",
alpha = 1
) +
scale_fill_manual(values = tp_colors) +
scale_shape_manual(values = c(
"D21" = 21, "T21" = 23
)) +
facet_wrap(~ panel, nrow = 1,
scales = "free") +
labs(x = NULL, y = NULL,
fill = "Timepoint", shape = "Genotype") +
guides(
fill = guide_legend(
override.aes = list(shape = 21)
)
) +
theme_pub(base_size = 10) +
theme(legend.position = "top")
```
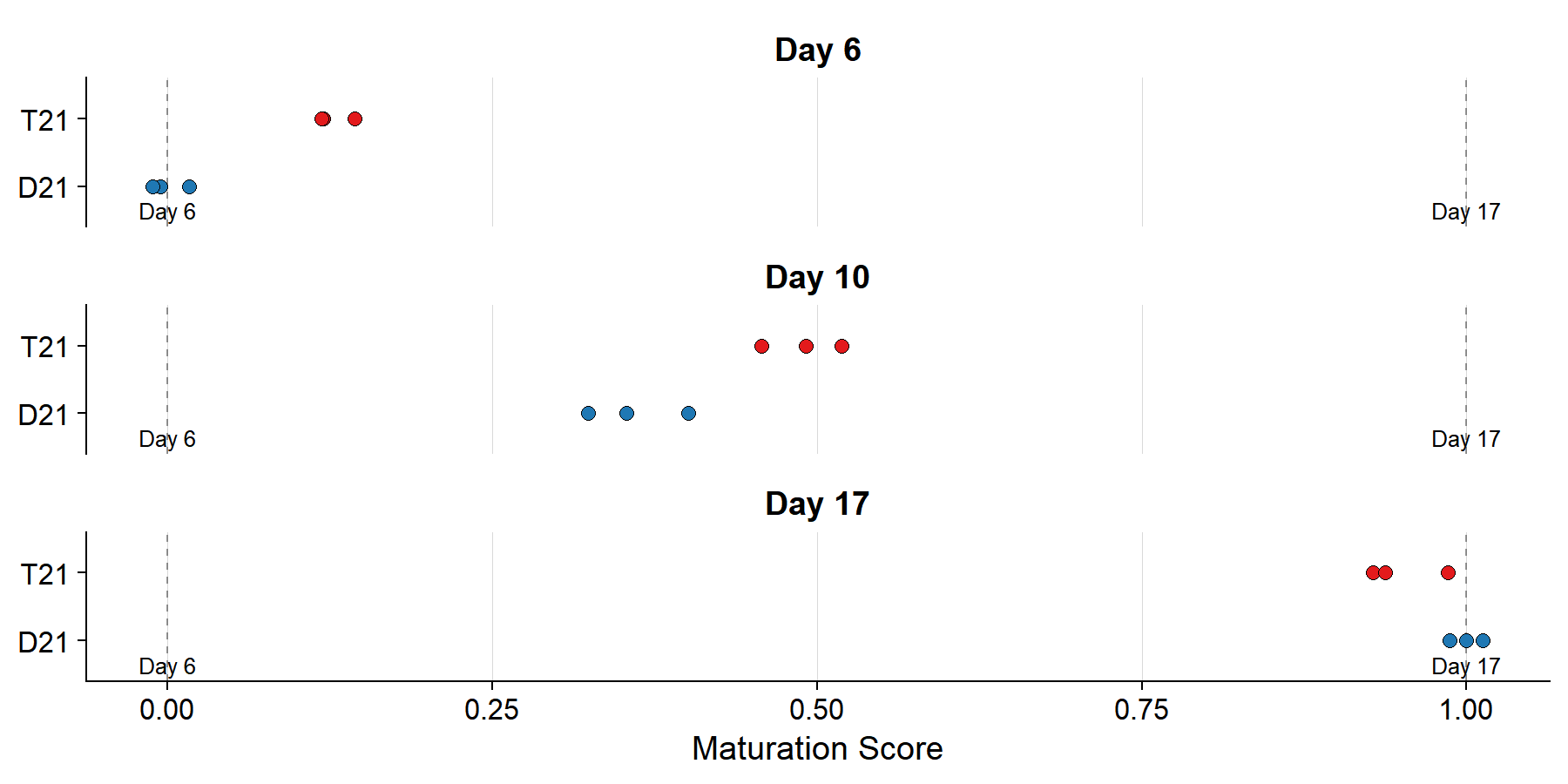
### Sample Projections onto Trajectory
```{r}
#| label: scores-compute
scores <- apply(all_proj[, 1:3], 1, function(z) {
sum((z - centroid_d6) * ref_vec) / sum(ref_vec^2)
})
score_df <- data.frame(
sample = meta$sample_id,
genotype = meta$genotype,
timepoint = factor(
paste0("Day ", meta$timepoint),
levels = c("Day 6", "Day 10", "Day 17")
),
score = scores
)
```
```{r}
#| label: fig-projection-strip
#| fig-cap: >
#| Each sample projected onto the reference vector.
#| Score 0 = Day 6 centroid, 1 = Day 17 centroid.
#| fig-height: 3
#| fig-width: 6
ggplot(
score_df,
aes(x = score, y = genotype, fill = genotype)
) +
geom_vline(
xintercept = c(0, 1),
linetype = "dashed", color = "#000",
linewidth = 0.3, alpha = 0.4
) +
geom_point(
shape = 21, size = 1.8,
stroke = 0.25, color = "black", alpha = 1
) +
facet_wrap(~ timepoint, ncol = 1) +
scale_fill_manual(values = c(
"D21" = col_d21, "T21" = col_t21
)) +
annotate(
"text", x = 0, y = 0.4,
label = "Day 6\ncentroid", size = 2.2,
hjust = 0.5, color = "#000"
) +
annotate(
"text", x = 1, y = 0.4,
label = "Day 17\ncentroid", size = 2.2,
hjust = 0.5, color = "#000"
) +
labs(x = "Maturation Score", y = NULL) +
theme_pub(base_size = 10) +
theme(
panel.grid.major.y = element_blank(),
legend.position = "none"
)
```
### Maturation Score Comparison
```{r}
#| label: score-table
score_summary <- score_df %>%
group_by(genotype, timepoint) %>%
summarize(
mean_score = round(mean(score), 3),
sd = round(sd(score), 3),
.groups = "drop"
)
knitr::kable(
score_summary,
col.names = c(
"Genotype", "Timepoint", "Mean Score", "SD"
),
caption = "Maturation scores by condition"
)
```
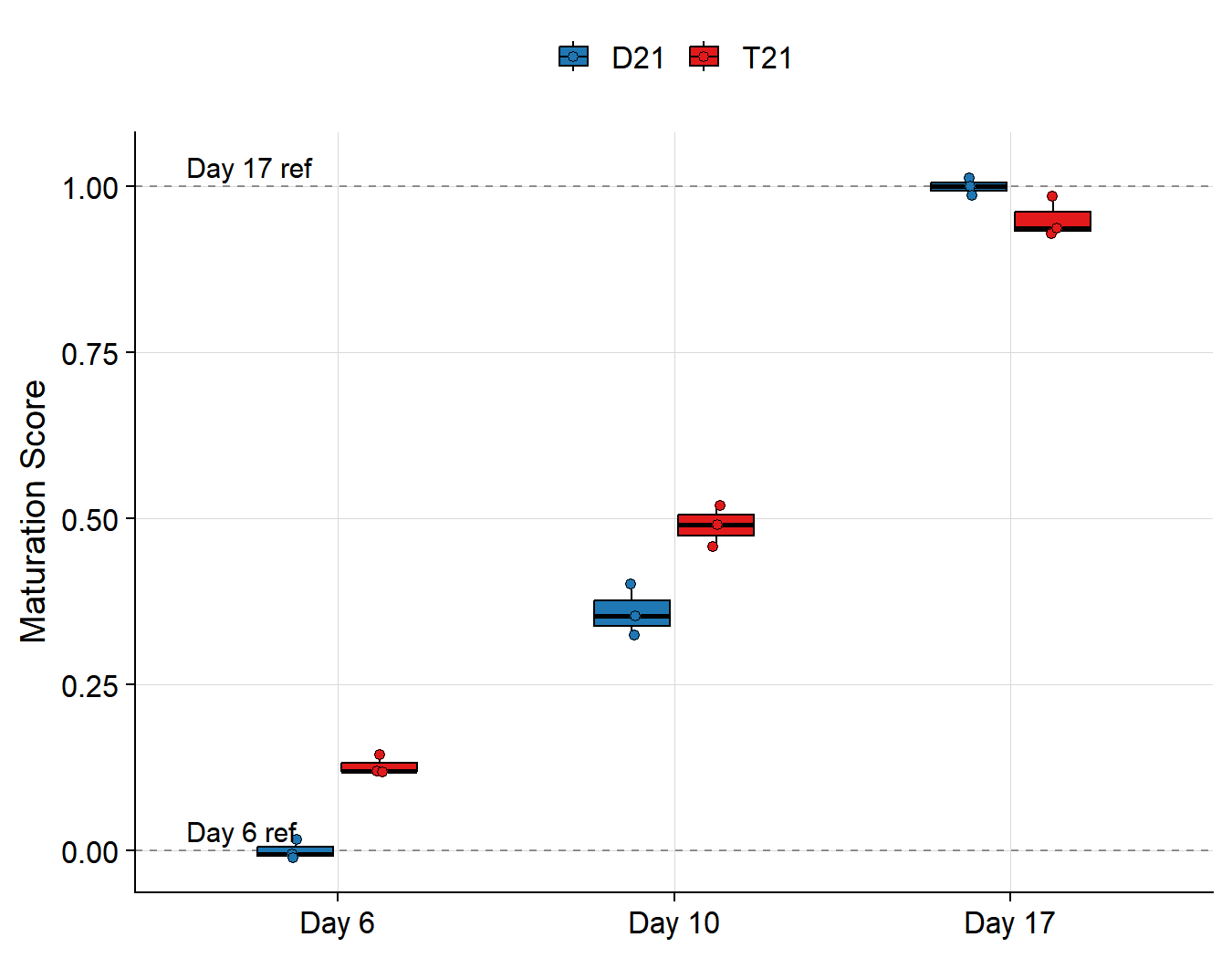
```{r}
#| label: fig-scores
#| fig-cap: >
#| Maturation score by genotype and timepoint.
#| Dashed lines mark Day 6 (s=0) and Day 17
#| (s=1) control centroids.
#| fig-height: 3.5
#| fig-width: 4.5
ggplot(
score_df,
aes(x = timepoint, y = score, fill = genotype)
) +
geom_hline(
yintercept = c(0, 1),
linetype = "dashed", color = "#000",
linewidth = 0.3, alpha = 0.4
) +
geom_boxplot(
width = 0.5, outlier.shape = NA,
alpha = 1, linewidth = 0.3,
color = "black"
) +
geom_point(
position = position_jitterdodge(
jitter.width = 0.05, dodge.width = 0.5
),
shape = 21, size = 1.2,
stroke = 0.25, color = "black",
alpha = 1,
aes(fill = genotype)
) +
scale_fill_manual(values = c(
"D21" = col_d21, "T21" = col_t21
)) +
scale_y_continuous(
breaks = seq(0, 1, 0.25),
expand = expansion(mult = c(0.05, 0.05))
) +
annotate(
"text", x = 0.55, y = 0.03,
label = "Day 6 ref", color = "#000",
size = 2.5, hjust = 0
) +
annotate(
"text", x = 0.55, y = 1.03,
label = "Day 17 ref", color = "#000",
size = 2.5, hjust = 0
) +
labs(x = NULL, y = "Maturation Score") +
theme_pub(base_size = 10) +
theme(legend.position = "top")
```
### Genotype Comparison
```{r}
#| label: comparison
d21_means <- score_summary %>%
filter(genotype == "D21") %>%
select(timepoint, d21 = mean_score)
t21_means <- score_summary %>%
filter(genotype == "T21") %>%
select(timepoint, t21 = mean_score)
diff_df <- inner_join(
d21_means, t21_means, by = "timepoint"
) %>%
mutate(
difference = round(t21 - d21, 3),
pct_diff = paste0(
round(difference * 100, 1), "%"
)
)
knitr::kable(
diff_df,
col.names = c(
"Timepoint", "D21 Score", "T21 Score",
"Difference", "% Difference"
),
caption = "Maturation score differences (T21 - D21)"
)
```
---
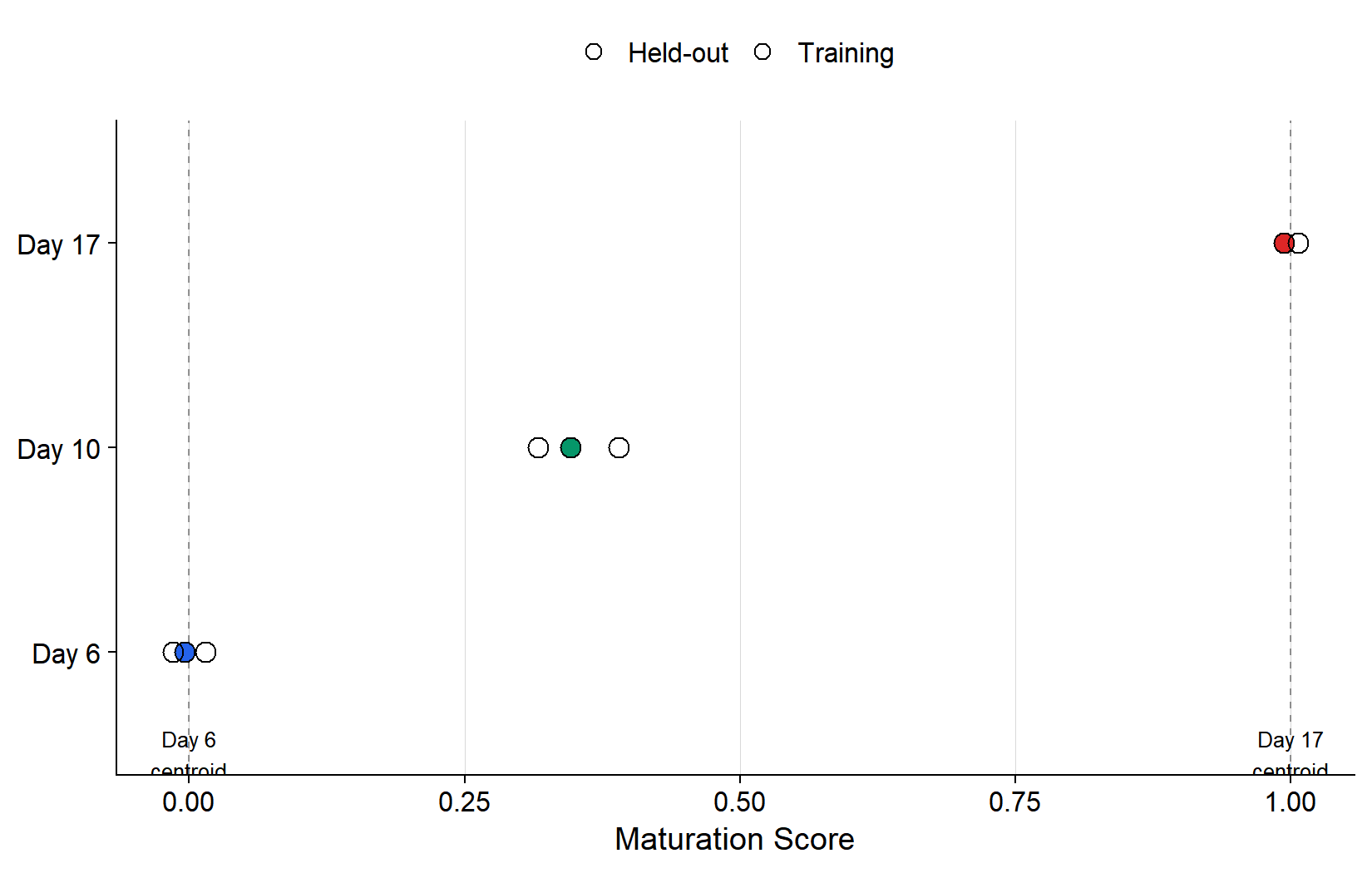
## Leave-One-Out Validation
To validate that the reference trajectory accurately
classifies samples by differentiation stage, we hold
out one replicate from each D21 timepoint, build the
trajectory from the remaining $n = 2$ replicates, and
score the held-out samples. No T21 samples are used.
```{r}
#| label: loo-compute
#| cache: true
# Hold out replicate 1 from each timepoint
holdout_ids <- c(
ctrl_meta$sample_id[
ctrl_meta$timepoint == 6][1],
ctrl_meta$sample_id[
ctrl_meta$timepoint == 10][1],
ctrl_meta$sample_id[
ctrl_meta$timepoint == 17][1]
)
train_ids <- setdiff(
ctrl_meta$sample_id, holdout_ids
)
cat(
"Training samples (n=6):",
paste(train_ids, collapse = ", "), "\n",
"Held-out samples (n=3):",
paste(holdout_ids, collapse = ", ")
)
# LRT on training set
train_meta <- ctrl_meta[
ctrl_meta$sample_id %in% train_ids, ]
train_counts <- raw_counts[, train_ids]
dds_loo <- DESeqDataSetFromMatrix(
countData = train_counts,
colData = train_meta,
design = ~ timepoint
)
dds_loo <- DESeq(
dds_loo, test = "LRT", reduced = ~ 1
)
res_loo <- results(dds_loo)
res_loo <- res_loo[order(res_loo$padj), ]
sig_loo <- rownames(res_loo)[
!is.na(res_loo$padj) & res_loo$padj < 0.05
]
n_top_loo <- min(1000, length(sig_loo))
top_loo <- head(rownames(res_loo), n_top_loo)
cat(
"\nLRT significant genes:", length(sig_loo),
"\nGenes used for PCA:", n_top_loo
)
# PCA on training samples
pca_loo <- prcomp(
t(as.matrix(vst_counts[top_loo, train_ids])),
scale. = TRUE, center = TRUE
)
# Project all D21 samples (train + holdout)
all_d21_proj <- predict(
pca_loo,
t(as.matrix(
vst_counts[top_loo, ctrl_meta$sample_id]
))
)
# Centroids from training samples only
tp_train <- as.numeric(
as.character(train_meta$timepoint)
)
c_d6 <- colMeans(
all_d21_proj[train_ids[tp_train == 6], 1:3]
)
c_d17 <- colMeans(
all_d21_proj[train_ids[tp_train == 17], 1:3]
)
rv <- c_d17 - c_d6
# Score all D21 samples
loo_scores <- apply(
all_d21_proj[, 1:3], 1, function(z) {
sum((z - c_d6) * rv) / sum(rv^2)
}
)
loo_df <- data.frame(
sample = ctrl_meta$sample_id,
timepoint = factor(
paste0("Day ", ctrl_meta$timepoint),
levels = c("Day 6", "Day 10", "Day 17")
),
score = loo_scores[ctrl_meta$sample_id],
set = ifelse(
ctrl_meta$sample_id %in% holdout_ids,
"Held-out", "Training"
)
)
```
```{r}
#| label: fig-loo
#| fig-cap: >
#| Leave-one-out validation. Reference trajectory
#| built from n=2 D21 replicates per timepoint
#| (open circles). Held-out replicates (filled)
#| are scored along the trajectory. Accurate
#| classification = held-out samples land near
#| their expected position.
#| fig-height: 3.5
#| fig-width: 5.5
ggplot(
loo_df,
aes(x = score, y = timepoint)
) +
geom_vline(
xintercept = c(0, 1),
linetype = "dashed", color = "#000",
linewidth = 0.3, alpha = 0.4
) +
geom_point(
aes(fill = timepoint, shape = set),
size = 2.5, stroke = 0.4, color = "black",
alpha = 1
) +
scale_fill_manual(values = tp_colors) +
scale_shape_manual(
values = c("Training" = 1, "Held-out" = 21)
) +
annotate(
"text", x = 0, y = 0.5,
label = "Day 6\ncentroid", size = 2.2,
hjust = 0.5, color = "#000"
) +
annotate(
"text", x = 1, y = 0.5,
label = "Day 17\ncentroid", size = 2.2,
hjust = 0.5, color = "#000"
) +
labs(
x = "Maturation Score", y = NULL,
shape = NULL, fill = NULL
) +
guides(
fill = "none",
shape = guide_legend(
override.aes = list(size = 2)
)
) +
theme_pub(base_size = 10) +
theme(
panel.grid.major.y = element_blank(),
legend.position = "top"
)
```
```{r}
#| label: loo-table
knitr::kable(
loo_df %>%
select(sample, timepoint, set, score) %>%
mutate(score = round(score, 3)) %>%
arrange(timepoint, set),
col.names = c(
"Sample", "Timepoint", "Set", "Score"
),
caption = paste(
"Leave-one-out validation scores.",
"Held-out samples should score near 0",
"(Day 6), ~0.5 (Day 10), and ~1 (Day 17)."
)
)
```
---
*Data: Martinez et al. (2024).
Front Cell Neurosci 18: 1341141.*